Post # 45: Technology of hydrogenation of edible oils for food uses – V : Complexity of the reaction, its implications and applications
Why is hydrogenation of oils so complex as a reaction?
All the usual ‘ingredients’ (and more!) making industrially conducted reactions complex are present:
- Series reactions: Where the product of a reaction is the reactant for another (in this case, similar) reaction.
Linoleic acid (9, 12 – cis,cis) → Oleic acid (9 – cis) →Stearic acid
- Parallel reactions: Where a reactant undergoes two simultaneous chemical transformations, the product of each of these can then undergo series and parallel reactions.
Linoleic acid → Oleic acid →stearic acid OR iso-oleic acid
→ Iso linoleic acid →Iso-oleic acid →stearic acid
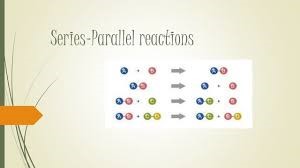
- Linoleic acid(9, 12 – cis,cis) →positional isomer, 9,13 –cis,cis
- Catalyst poisoning: The usual suspects are the residual phosphatides and suplhur compounds. They slow down hydrogenation per se’ thru enhanced isomerization and reduced hydrogenation episodes at the catalyst surface simply thru reduction of active sites.
- Non-hydrogenation glyceride reactions: Mainly hydrolysis of the ester linkages, thermal breakdown and limited fa rearrangements.
- Non-glyceride reactions: Mainly isomerization and hydrogenation of pigments.
- The unclassical use of IV as the concentration indicator. Even the corrected [db], mols/lit, failing to represent the fact of double bonds being sporadically distributed parts of large molecules in the sense that [db] tends to denote mols/lit of hypothetical, free, uniformly free-floating double bonds. And then, [db] = [cdb] + [tdb]. Unsatisfactorily defined kinetics in terms of (i) molecularity and order (ii) IV vs [db] (iii) no cognizance of the changing molecular orientation, shape and size factors captured by Arrhenius Constant ‘A’ and (iv) varying ∆Ea. Note that real commercial process is not isothermal, at least in the starting large extent, hence interpreting the observed kinetics during such a stage becomes a nightmare.
Hydrogenation – a unique model for a complex ‘multiphase’ reaction. Chemical Reaction Engineering is a specialized and elegant branch of Chemical Engineering focused on deep dives in defining reaction rates, the interrelationship between reaction mechanisms and their ‘rate equation’, the non-concentration factors determining the rates and how ‘mass transfer phenomena’, complicate their intrinsic rates. Such complications can stem from the physical states (‘phases’) of the reactants (and the catalyst, if there is one), presence of catalyst poisons and presence of series and parallel reactions. Food scientists and technologists, are usually not familiar with this interesting branch of chemical engineering. But ‘hydrogenation of edible oils for food uses’ is an excellent opportunity to familiarize them with these reaction intricacies that they can’t disown because of their food uses!
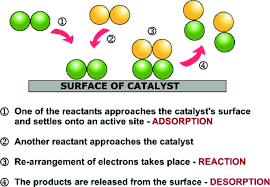
You will easily see how Nickel catalysed hydrogenation of edible oils for food uses is a classical ‘Heterogeneously catalysed multiphase reaction’ but with complexities thrown in. Let’s dive deeper into its usual and distinguishing features:
- A two phase reaction in terms of reactants. The bulk reactant – the oil – is liquid but the other reactant – hydrogen gas – is dispersed in it both as bubbles and in dissolved form. Fascinating drama plays out as the bubbles rise while the gas diffuses into the reaction mixture and thence to the catalyst surface and finally into the crevices or channels to reach the active sites for a rendezvous with good old double bonds!
We will have to bypass the fascinating, interconnected drama played out among gas inlet rate, reaction rate, headspace pressure ‘p’, temperature rise because of the reaction exortherm and [Hyd], mols/lit. (Reaction mixture temperature ‘T’ and ‘p’ together increase hydrogen solubility and hence [Hyd], mols/lit). For the record, this is my limitation as a Food Technologist who is not a full-fledged Chemical Engineer. Thus the oil as a continuous liquid reactant is in reactive contact with a gas. Obviously, the limited solubility of hydrogen in oil is an insurmountable obstacle that queers the relative proportions of the double bonds and hydrogen gas approaching the active catalyst sites. In isobaric, isothermal hydrogenation, the [db]/[Hyd] ratio must ‘invert’ at a calculatable IV – a loaded observation implying scope for more meaningful reaction steering.
Obviously, hydrogen diffuses continuously from the bubbles and the headspace into the oil as long as hydrogen concentration gradient is from the bubble/headspace to the catalyst sites thru a robust hydrogenation rate.
- Heterogeneous catalysis: The catalyst is a solid i.e. heterogeneous to the reaction mixture and this makes it a ‘heterogeneously catalysed multi-phase reaction’.
- The series and parallel reactions and their implications: Apart from the most unsaturated PUFA (with all cis double bonds) pre-existing in the original oil, all the oil molecular entities in the reaction mixture at any ∆I during hydrogenation, can be either pre-existing or be just formed by reaction. For example, all 18 carbon-single unsaturation fatty acid at any ∆I during hydrogenation can be…..
– unreacted portion of original cis oleic acid,
– only isomerized part of original oleic acid, and
– any isomer of C-18, single unsaturation fatty acid formed by hydrogenation of linoleic acid which itself may be original unreacted cis-cis fatty acid or a product of hydrogenation of linolenic acid or isomerized linoleic acid.
This extreme complexity is the result of simultaneous series and parallel reactions occurring at all stages of hydrogenation. In the former, PUFA becomes MUFA which becomes SFA. In the latter, a PUFA becomes an isomeric PUFA as well as MUFA simultaneously. This simultaneous transformation of single molecular entities into several entities is a wonderful tool to understand/teach series and parallel reactions. After hydrogenation, filtration separates the catalyst from the reaction mixture and the filtrate, as a whole, is the product.
The implications of the complexity of hydrogenation of oils
Implications of reaction pathways: From oxidation-resistance stand point, we would like to preferentially hydrogenate PUFA i.e. ensure that their ‘concentration’ is sufficiently reduced (with ideally, linolenic acid reduced to < 2 % so that it is available as an efa but its oxidation vulnerability is dampened) before SFA generation from MUFA is fast. Thus the product will have a near perfect combination of oxidation resistance and ffa profile, for given I. This obviously translates to the tough task of not allowing the PUFA hydrogenation to slow down significantly, despite its quickly diminishing ‘concentration’, in the face of the usually high MUFA concentration.
As we have noted earlier, this requires the hydrogen supply at catalyst sites to be limited so that PUFA hydrogenation is spurred by their higher reactivity rather than hydrogen supply. This inevitably promotes of trans (TFA) formation. (In fact, modulating hydrogen concentration, [Hyd], mols/lit, in consonance with the stage of hydrogenation in case of any oil, can be a powerful tool in controlling the quality and rate of hydrogenation.)
The hydrogen-abundant indiscriminate hydrogenation has the bulldozing effect of series fatty acid hydrogenation (relentless reduction in PUFA to MUFA and MUFA to SFA). Here, MUFA (with higher concentration compensating for lower reactivity) hydrogenation competes with PUFA and allows some PUFA to linger when significant amounts of new SFA have been formed from MUFA for any ∆I. Thus for the same product IV as in the previous case, there is more liquid at any temperature and poor oxidation-resistance. (In terms of oxidation resistance: SFA >> MUFA > PUFA.)
Obviously, such a product, if solidified by cooling, will melt (or ‘collapse’) fast i.e. be quick-melting i.e. have a steep melting profile. Reminds you of moulded bar chocolate? But with two important side effects: TFA reduction is better but oxidation sensitivity, worse as a result of lingering PUFA. Win some, lose some, says nature.
What is the reactant? What is [R], mols/lit?: The main bulk reactant is a natural product (and not sodium chloride or benzene) with its own variations. Actually, for any hydrogenation feedstock, it is bewildering to decide what the actual reactant is: the double bond or the unsaturated fatty acid or the oil molecule as a whole? Ironically, the sheer convenience of Iodine Value as an indicator of the rate of hydrogenation (∆I/∆t) and of the extent of hydrogenation (∆I = Io – I) has served to fix the double bond as the reactant. But in a non-classical, non-kinetics compliant way!
Interestingly, imaginary (!) isolated double bonds (>C=C<) would drift into catalyst channels (or crevices) at rates comparable with hydrogen but not huge oil molecules! In other words, double bonds as a mass fraction of oil are fine as unsaturation indicators, but they are slowed down by a lot of ‘dead weight’! Vastly different from hydrogenating ethylene to ethane!
Research/education tool in chemical engineering and food technology
The end result of this ‘set of reactions’ called hydrogenation of edible oils is the reaction mixture containing the hydrogenated product and the ‘spent’ (but still reusable) catalyst. Obviously, the hot liquid separated from the catalyst thru simple filtration is the product that, in most cases, cannot bear the cost of any separation processes as in most pure chemical reactions. And all the effects of IV reduction and isomerizations are hidden in it which its lab analysis immediately reveals giving it its functionality. But the essence of the physical and chemical drama played out is the understanding of the various ‘resistances’ to the progress of the reaction.
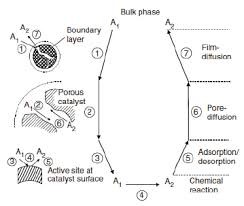
– the double bonds (as parts of elaborate oil molecules) and hydrogen have to diffuse (swim) thru the reaction mixture thru the crevices of catalyst particles and reach the active catalyst sites which amounts to a ‘physical’ resistance prior to the reaction per se’
– the ‘just prior’ set of reactants from each site has to vacate it for the next set and quickly emerge from the catalyst particle channel with minimal jostling with the in-coming sets
– the inherent, inevitable resistance in terms of overcoming the reaction’s in-built Energy of Activation (Ea) barrier to pass into hydrogenated product decreases with rise in temperature as the great Svante Arrhenius explained over a century ago. This rate enhancing effect, obviously during the ‘rising temperature stage’ in commercial hydrogenation, is countered by quickly reducing ‘reactant concentration’. Thus, in dI/dt or d[db]/dt = [A.e^∆Ea/RT]*I or [db], the net or observed rate is the result of two conflicting influences on the rate. This realization is loaded with research avenues.
– the ∆Ea on [db] basis is rising with the build up of TFA which is another factor dampening the rate enhancement because of temperature rise.
Important practical points to note:
- how reaction conditions determine the product quality thru their influence on the aforesaid resistances
- there are avenues for ‘managing’ the reaction with the progress of time or ∆I or instantaneous IV.
- how several fresh graphical plots can be ‘spun off’ from standard ones for fresh insights and
- catalysis as a surface phenomenon
- the unique ‘reaction conditions – reaction trajectory – product quality – product functionality – processed/cooked food experience’. It would be a new experience, especially for chemical engineers, to realize that the same reactants in the same reactor can deliver two different food consumption experiences depending on reaction conditions that determine reaction trajectory.
Next Post:
The treatment of hydrogenation kinetics in mainstream literature
Well-meant simplifications complicate the narrative and mask research avenues
Visit Disclaimer