Post #46: The treatment of hydrogenation kinetics in mainstream literature – Well-meant simplifications complicate the narrative and are unclassical
The ocean of literature created as a result of ‘research’ in hydrogenation of edible oils for food uses is daunting. We had a glimpse of a purposeful version of that in post no. 42, Hydrogenation of edible oils for food uses : How it evolved and where it stands today. However, research in a resourceful environment, seems to have a momentum and branching of its own, creating a bewlildering (admittedly, for this author) labyrinth. Inter alia, this seems to have mandated simplifications in literature aimed at a cross-section of the society.
The success of such simplification has been impressive and this author admits to have benefited from it; in any case, there can never be an argument against it. But it throws up opacities in the understanding of the observed kinetics and trajectory of the reaction. They served their purpose of helping reach vast swathes of students, marketers, researchers and academics, by ‘talking in their language’. But didn’t it also present all-pervading, bewildering deviations form ‘first order, monomolecular kinetics’ without proper rationalizing?
 ‘Nothing is so good that it cannot be made better’, right? This post is a humble attempt at doing that. The idea is to put the mainstream treatment of hydrogenation kinetics under a microscope, try to determine where there is need for and possibilities of adopting more rigorous kinetic treatment and thereby induct a larger cross-section of researchers – especially chemical engineers. A collateral benefit can be the wisdom that can lead to more meaningful steering of the commercial process.
‘Nothing is so good that it cannot be made better’, right? This post is a humble attempt at doing that. The idea is to put the mainstream treatment of hydrogenation kinetics under a microscope, try to determine where there is need for and possibilities of adopting more rigorous kinetic treatment and thereby induct a larger cross-section of researchers – especially chemical engineers. A collateral benefit can be the wisdom that can lead to more meaningful steering of the commercial process.
Simplified kinetics in mainstream literature
In chemical reaction kinetics, concentration of reactants/products expressed as [R], mols/lit and [P], mols/lit is sacrosanct barring rare exceptions. This stems from the importance of the spatial distribution of reactants (and products) in the reaction mixture which helps determine their effective collision frequencies at any temperature. This is obviously the non-energetics part of the rate law; the energetic part comes from the famous Arrhenius equation for rate constant ‘k’. Now note the following less noticed complications in case of hydrogenation of oils:
- What’s the real reactant?: The actual reactant are the double bonds on the fatty acid chains which
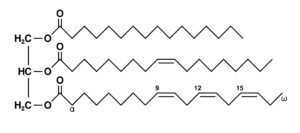
Triglyceride are esterified with the 3 –OH groups of glycerol. These are merely unevenly distributed ‘spots’ on some of the oil molecules which, being large, make it look like sparse garnish on a large surface of a dish. Classical kinetics recognizes only molecules, atoms, ions and free radicals as reactants to factor in the shape and size effects determining the collision effectiveness (as well as diffusion coefficients.) Cholesterol or beta carotene molecules have smaller
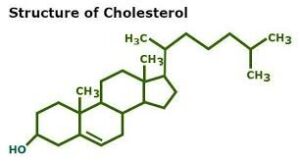 sizes but they have fairly rigid ring structures or branches. Hence a small functional group on them would be pretty much ‘fixed’ and hence its kinetics would be predictable in terms of A and Ea in the Arrhenius equation for the k-∆Ea relationship. In their case, an analytical parameter (like IV in case of oils) indicating the concentration of the functional group would meaningfully indicate the concentration of the macromolecule, especially if expressed
sizes but they have fairly rigid ring structures or branches. Hence a small functional group on them would be pretty much ‘fixed’ and hence its kinetics would be predictable in terms of A and Ea in the Arrhenius equation for the k-∆Ea relationship. In their case, an analytical parameter (like IV in case of oils) indicating the concentration of the functional group would meaningfully indicate the concentration of the macromolecule, especially if expressed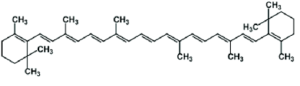
Beta carotene in mols/lit units.
- The diverse products: This is further complicated by the generation of numerous molecular entities during hydrogenation and isomerization. Of these, trans isomers have more enthalpy (greater negative value) stemming from the inherent extra stability of ‘straightened’ double bonds – mimicking the single sp3-sp3 sigma bonds. This obviously means higher Ea of hydrogenation (and lower ∆Hr) and corresponding slowing of the reaction for the same IV. This diversity of the ‘reactant’ at the start and complicating with the progress of the reaction is a huge energetics reason for the bewildering kinetics.
- IV as ‘concentration’ indicator and kinetics fulcrum: Undoubtedly, IV was too convenient as a ‘quantitative indicator of unsaturation or double bonds’ to be ignored. And the sustained success in churning out hydrogenated stocks with IV as reaction progress indicator could have brushed aside murmured objections of classical kineticists. But the fact is: IV is a mass based indicator of unsaturation while classical kinetics recognizes only molecules, atoms and free radicals as reactants expressed in ‘concentration’ as [R], mols/lit.
Focusing on the reaction mixture, its density decreases continuously during hydrogenation for both physical and chemical reasons (slowing down during the isothermal stage from disappearance of the physical reason). Hence IV 100 would mean two different concentrations at 140 and 190 deg C in terms of concentration, though not in a major way. From eq. 1, [db] mols/lit = 0.0394ρI, where ρ and I are the instantaneous density of the reaction mixture and IV of the oil during hydrogenation.
If the kinetics were first order, monomolecular:
d[db]/dt = k’[db] at a constant temperature. For the rising temperature stage of commercial hydrogenation, k’ would vary (increase) continuously offsetting the effect of diminishing reactant concentration. At least conceptually, a stage should come when the rate should appear practically constant if temp is still rising and [db] has reduced significantly.
Idρ/dt + ρdI/dt = k’ρI or dI/dt = k’I – (I/ρ)(dρ/dt)……………………2
- The molecularity and the order of the reaction: The ‘first order’ was assigned to the reaction logically; ‘the rate had to be directly proportional to the reactant concentration’. However, the literature is replete with kinetic plots of hydrogenation batches which follow first order kinetics only in rare exceptions, deviation (sometimes wide) is the rule. Some of those stem from sticking to treating hydrogenation as a monomolecular, first order reaction. This has been pointed out in the previous post.
Thus the bewildering, all-pervasive deviations (accepted with somewhat yogic equanimity) in observed hydrogenation kinetics stem from…
- not recognizing the flaws in IV as concentration indicator
- ignoring the effect of hydrogen concentration, [Hyd], mols/lit which is constant only in ‘contrived’ isothermal, isobaric hydrogenation of the lab. Interestringly, constant [Hyd] means hydrogen input rate automatically reducing with increasing ∆I.
- not recognizing the variations in ‘k’ even at constant temperature
- failure to accurately ‘mass balance’ hydrogen to determine ‘IV-reducing hydrogen consumption’. This point is elaborated in the next post describing the innovation to know instantaneous IV during hydrogenation, uninterruptedly.
The IV vs [db], mols/lit issue and its solution
The density of an oil varies with temperature on expected lines. It also varies with the average molecular weight of the constituent fatty acids and the IV of the oil. But these are all ‘narrow band’ variations. Obviously, any specific oil would change its density only with temperature in absence of a chemical reaction. During commercial hydrogenation, the temperature rises from the starting minimum (typically 120 deg C), is held constant at a pre-decided higher temperature (typically 180 deg C) and held there through cooling water circulation to offset the reaction exotherm.
This is pretty much the SOP. (Don’t ask me how we managed during my days in absence of temperature transmitter controlling cooling water flow; I have opened the cw valve myself many times, sometimes followed by pretty unsettling water hammer! Additionally, there is finely dispersed Ni catalyst. Thus even, the bubble-less reaction mixture has a specific density to start with which changes during the course of hydrogenation; obviously only thru chemical changes during the isothermal stage and hence limited. Thus, the reaction mixture is a unique material with its unique variation pattern.
All this fuss is about migrating to [db] from IV and place hydrogenation on the common kinetic platform to invite chemical engineers (which we food technologists are not!) to take a closer look first at constant condition hydrogenation and then conventional hydrogenation incorporating rising and constant T spells. Eventually, of course, we should be migrating to constantly modulated conditions for the best optimum between rate and trajectory or time-productivity and quality of product. In fact this is a big pointer to the potential of cross-discipline collaborations. Chemical engineers would be delighted to connect their knowledge with decreasing TFA or producing highly economical CLA as a part of cooking oil.
A protocol to derive insights in isothermal hydrogenation:
Break the ∆I into several small ∆I1, ∆I2 + ∆I3……
Convert each spell into ∆[db].
Derive d[db]/dt vs t, [db] curves for each ∆db using chemical hydrogen consumption rates. More about it later.
Look for pseudo-zeroeth order in the first ∆[db] for high IV oils.
Break later ∆[db]’s into a series of ‘pseudo first order spells’ to arrive at a series of ‘k’ values. Conclude that variation in k mean that changes in A and ∆Ea are failing to cancel each other.
Work out the constantly decreasing [db] or [db]/[Hyd] values. Appreciate its effect on each ∆[db] spell.
Compare k values at the start (when [tdb] is zero), when [tdb] is likely to be maximum and when it has declined sufficiently. Note that the [tdb] maxima which cannot be far from the k maxima given the known difference between the enthalpies of hydrogenation of cis trans double bonds.
Mainstream literature
Bailey’s ‘volumes’ have been bibles to generations of food technologists and will continue to reassure coming generations merely thru their presence on the tables and bookcases. Personally, I think that people like Bailey and Patterson (and others like Octave Levenspiel) deserve the same status in the world of Science and Technology that Abraham Lincoln, Mahatma Gandhi and Nelson Mandela have in social and political life. Post independence and slavery elimination, where would we be without the enlightening benevolence of these greats?
I am convinced that I would not be able to write that chapter on Hydrogenation in Baliey’s; I wouldn’t know what to include and what to exclude, for starters. And how to weave the ‘inclusions’ into a coherent narrative. I hereby thank its author; I know what it must have taken. But I wish the IV-based, first order monomolecular kinetics had been explained as a ‘convenient’ tool and the mass transfer phenomena explained in detail matching their relevance. With them, the rampant and bewildering deviations in hydrogenation rates and trajectories would have looked less mysterious and conceivably attracted chemical kineticists to till this fertile land. Who knows, this, combined with the involvement of ‘those magicians called catalyst makers’ would have saved hydrogenation from its eventual fate!

 For a food technologist like me with my limited vision and knowledge, mainstream hydrogenation literature is that chapter in Bailey’s volumes and the book, ‘Hydrogenation of Oils and Fats’ by HBW Patterson. I daresay: most people on this planet are limited to these two resources. If Mr. Patterson is alive today, I would like to fly over just to: (i) understand how he managed to pack such a book in a lifetime, (ii) to thank him for including the mass transfer phenomena in his discussions, (iii) understand his ‘equation for solubility of hydrogen in most oils’ to derive my own equation for calculation of [Hyd], mols/lit at any p,T, and to tell him that (iv) he has in me, a student of this elegant modification technique who will never be able to match his awesome, sweeping grasp of it.
For a food technologist like me with my limited vision and knowledge, mainstream hydrogenation literature is that chapter in Bailey’s volumes and the book, ‘Hydrogenation of Oils and Fats’ by HBW Patterson. I daresay: most people on this planet are limited to these two resources. If Mr. Patterson is alive today, I would like to fly over just to: (i) understand how he managed to pack such a book in a lifetime, (ii) to thank him for including the mass transfer phenomena in his discussions, (iii) understand his ‘equation for solubility of hydrogen in most oils’ to derive my own equation for calculation of [Hyd], mols/lit at any p,T, and to tell him that (iv) he has in me, a student of this elegant modification technique who will never be able to match his awesome, sweeping grasp of it.
Next Post:
Adaptating the commercial reactor for research – I
How imagination midwifes innovation: Headspace gas quantification
Visit Disclaimer